

|
|
Hotel Alcora
Ctra. de San Juan de Aznalfarache -
Tomares, km 1
41920 SEVILLA
[Objetivo] [Áreas] [Programa] [Agenda] [Comités] [Entidades] [Comunicaciones] [Inscripción] [Boletín de Inscripción]
El diseńo de fármacos por ordenador
D. Víctor Segarra Matamoros
Departamento de Diseńo Molecular. ALMIRALL
PRODESFARMA
Introducción
De entre las nuevas metodologías emergidas recientemente, cabe destacar el Diseńo de Medicamentos Asistido por Ordenador. La Modelización Molecular ha mostrado su gran aplicación en el campo de la Química Terapéutica, siendo hoy día un instrumento básico en la Investigación Farmacéutica.
Las diferentes técnicas de Química Computacional y Modelización Molecular contribuyen de forma eficiente en las distintas etapas para la propuesta de nuevos fármacos.
Descubrimiento de nuevos ‘cabezas de serie’
Una vez definido el objetivo terapéutico y el proceso molecular o mecanismo de acción involucrado, la identificación de nuevos compuestos con actividad biológica destacada y estructura química novedosa (denominados ‘leads’ o ‘cabezas de serie’) constituye uno de los primeros pasos en el descubrimiento de nuevos fármacos.
Los avances en el campo de la Genómica y de la Biología Molecular, han incrementado en gran número los nuevos objetivos biológicos de interés. La gran capacidad de automatización, tanto en la preparación de bibliotecas químicas (Química Combinatoria, Química de Alta Eficacia) como de su rápida evaluación biológica (HTS: screening de alto rendimiento), han permitido el ensayo de un gran número de compuestos en los nuevos mecanismos identificados, los denominados screening masivos o aleatorios.
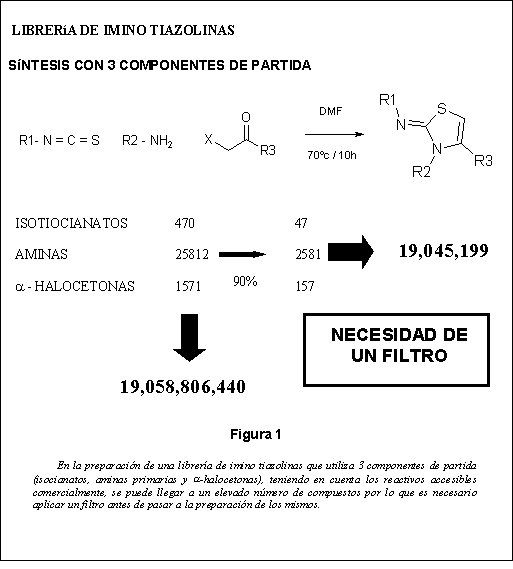
No obstante, tal como se muestra en el ejemplo de la figura 1, al plantear el diseńo de una biblioteca de productos químicos basado en una determinada síntesis, se pueden obtener un gran número de compuestos1. En dicho ejemplo teniendo en cuenta los reactivos accesibles comercialmente2 se podría llegar hasta un total de 19 mil millones de compuestos. Incluso aplicando una reducción del 90% para cada una de la series de reactivos se llegaría a 19 millones de compuestos. Esto hace que sea necesario aplicar un filtro con la finalidad de decidir qué compuestos hay que sintetizar3-6.
Cuando el objetivo de la biblioteca es el descubrimiento de nuevas moléculas cabezas de serie, tiene sentido pensar que la diversidad entre los compuestos seleccionados esté relacionada con la capacidad de interaccionar con la nueva diana biológica (receptor, enzima, DNA), y que por tanto, puedan contener el mayor número posible de farmacóforos.
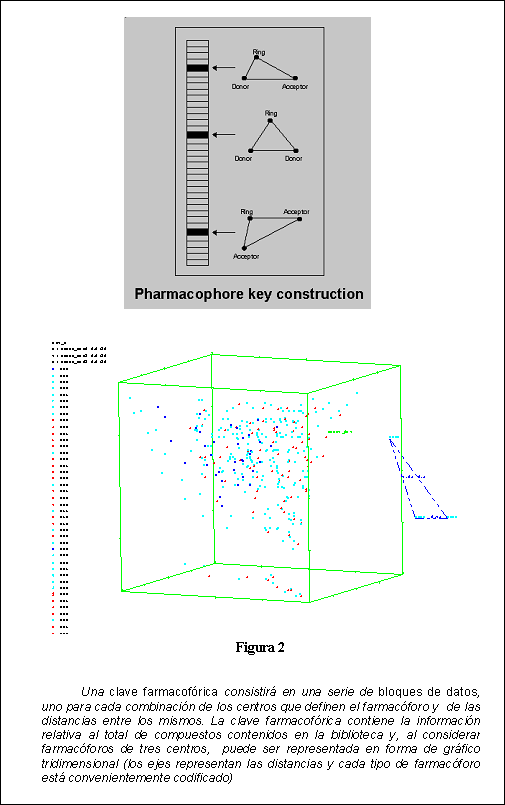
Basándose en este concepto, se ha desarrollado la denominada Diversidad en Función del Farmacóforo5-7 que se basa en generar una serie de claves farmacofóricas (figura 2) que contienen la información de la descripción del farmacóforo en función de los tipos de centro y las distancias entre los centros. De esta manera los distintos compuestos si aportan nuevos tipos de farmacóforos serán seleccionados para su preparación, mientras que, en caso contrario, serán excluidos. Los farmacóforos considerados pueden ser de 3 ó 4 centros. Finalmente, la clave farmacofórica para un determinado grupo de moléculas (teniendo en cuenta la flexibilidad conformacional) se puede representar en un gráfico tridimensional (figura 4) codificado en función de los tipos de farmácoforo e indicando las distancias entre los centros en cada uno de lo ejes de coordenadas (para farmacóforos de tres centros). Cada uno de los puntos representados puede ser seleccionado obteniéndose el farmacóforo en particular.
Expansión de nuevos ‘cabezas de serie’
A partir de los estudios de modelización molecular indirecta (basados en la estructura tridimensional de los ligandos) y modelización molecular directa (basados en la estructura tridimensional de la macromolécula diana terapéutica como el receptor, enzima o DNA) se obtienen los requerimientos básicos para una determinada actividad, es decir, lo que entendemos por farmacóforo (conjunto de propiedades estructurales, fisicoquímicas y electrónicas, básicas para una determinada actividad farmacológica).
Por tanto, en esta etapa se buscarán nuevas familias químicas que coincidan con estos requisitos plasmados en el farmacóforo.
En este sentido, una de las estrategias seguidas con mayor éxito es la búsqueda de estos farmacóforos en diversas bases de compuestos.
Búsqueda del farmacóforo en Bases de Datos Tridimensionales
El objetivo fundamental de las búsquedas de fármacoforo en bases de datos tridimensionales consiste en la identificación de estructuras antiguas (sintetizadas previamente para una actividad biológica distinta o pertenecientes a un catálogo comercial) pero que encajan con los requisitos necesarios (farmacóforo) para la actividad biológica objeto actual de interés.
Las bases de datos 3D no sólo contienen las estructuras de los compuestos con las coordenadas en disposición tridimensional, sino que además están parametrizadas de forma que se puedan llevar a cabo búsquedas de farmacóforos.
En la parametrización de estas bases de compuestos se deberá incluir la descripción de los posibles centros de interacción (centros aceptores y dadores de puentes de hidrógeno, centros hidrofóbicos, núcleos aromáticos, centros que pueden actuar como ácido o base, etc). Al realizar la búsqueda del farmacóforo se deberá tener en cuenta también la flexibilidad de las moléculas, y por tanto, deberán ser consideradas diversas conformaciones.
Tradicionalmente, estas bases de compuestos podían ser corporativas (conteniendo la colección de compuestos sintetizados por las compańías a lo largo de los ańos) o comerciales (BioByte, Derwent, Maybridge,..). Actualmente, a estas fuentes de compuestos se les han ańadido las grandes bibliotecas provenientes de la Química Combinatoria y Química de Alta Eficacia en general. Además, este tipo de búsqueda permite ser realizado no sólo sobre las bibliotecas de compuestos ya sintetizados sino sobre bibliotecas virtuales con un elevado número de compuestos que pueden no haber sido sintetizados pero de los cuales se dispone de una manera rápida de prepararlos y la accesibilidad de sus reactivos.
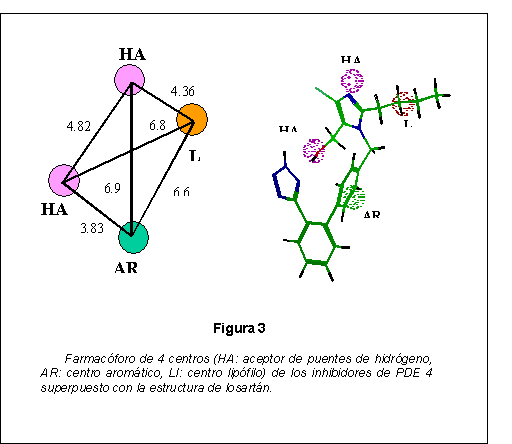
A modo de ejemplo, en el campo de los inhibidores de la fosfodiesterasa 4 (PDE 4) se ha descrito recientemente un farmacóforo relacionado con un determinado grupo estructural de inhibidores8. A partir de un fármacóforo simplificado de 4 centros se ha realizado la búsqueda del mismo en diversas bases de datos tridimensionales9. De entre los compuestos seleccionados en dicha búsqueda (figura 3) cabe destacar el losartán, compuesto antihipertensivo que actúa como antagonista de la Angiotensina II. La evaluación biológica posterior de este compuesto ha mostrado que, a pesar de no ser muy potente, es activo en la inhibición de la PDE 4.
Optimización de cabezas de serie
Tras la identificación de un compuesto, perteneciente a un grupo químico novedoso, y con una actividad biológica destacable, el siguiente paso consistirá en la introducción de pequeńos cambios en su estructura con la finalidad de mejorar la potencia, selectividad, comportamiento farmacocinético, tasa de metabolismo, etc.
En esta fase, donde se pueden llegar a preparar un gran número de derivados, presentan gran interés los estudios cuantitativos de estructura-actividad (QSAR). En lo estudios QSAR se correlaciona la actividad biológica con un gran número de propiedades de la molécula (fisico-químicas, electrónicas, estructurales), identificándose las más relevantes.
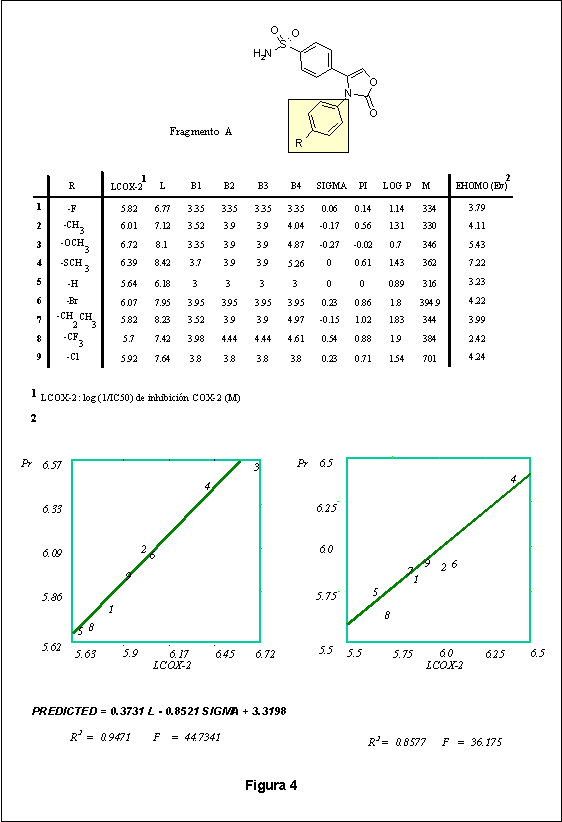
En la figura 4 se observa un ejemplo10 en el cual se correlaciona la actividad inhibitoria de la ciclooxigenasa-2 (COX 2) con diversas propiedades, tanto estructurales (como el parámetro L de Verloop, relacionado con la longitud del substituyente) como electrónicas (el parámetro sigma y la energía del orbital frontera HOMO)
Referencias
[Objetivo] [Áreas] [Programa] [Agenda] [Comités] [Entidades] [Comunicaciones] [Inscripción] [Boletín de Inscripción]

[Actividades de la SEIS] [Revista
I+S] [Solicitud de Inscripción]
Secretaría Técnica de Inforfarma'99:
CEFIC
C/ Olimpo, 33, 1ş C
28043 - Madrid
Telfs: (91) 388 94 78 / 79
Fax: (91) 388 94 79