| Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
Santander, 15 de octubre de 2004
HOSPITAL UNIVERSITARIO MARQUÉS DE VALDECILLA
SantanderDra. Mª Luisa Cagigal Cobo
TUMOR FIBROSO SOLITARIO MALIGNO DE LA PLEURA
Resumen de la historia clínica
Mujer de 55 años con dolor costal izquierdo de características pleuríticas. En TAC se observa una tumoración pulmonar izquierda con áreas de necrosis. Se practica extirpación de la masa
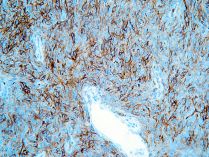
Diagnóstico
Tumor fibroso solitario maligno de la pleura
Comentario
Os presentamos el caso de una mujer de 55 años, que como único antecedente de interés, padece una hipertensión arterial tratada farmacológicamente.
No existen antecedentes de exposición al asbesto ni tabaquismo.
La paciente consulta por un dolor costal izdo, de características pleuríticas, de varios días de evolución, que no se acompaña de tos, disnea ni fiebre.
Se le realiza una radiografía de tórax, donde se objetiva una masa en pulmón izdo y un derrame pleural ipsilateral , que desaparece en un control posterior.
Así mismo, se le realiza un TAC con contraste que es informado como masa de 6,5 cm de diámetro en el segmento 6 del lóbulo inferior del pulmón izdo, que se realiza con contraste y que pressenta áreas hipodensas en su interior sugestivas de necrosis.
Se punciona dicha masa y el diagnóstico de la punción-aspiración es de tumor fibroso pleural .
A la paciente se le realiza una tóracotomía con extirpación del tumor, informando los cirujanos de su localización extrapulmonar.
En nuestro departamento recibimos una tumoración de 7 cm de diámetro mayor, encapsulada , con un patrón vascular prominente y no pediculada. No se objetivó tejido pulmonar asociado.
Dicha masa era sólida, firme, de coloración blanquecina, con un puntedo amarillento focal .
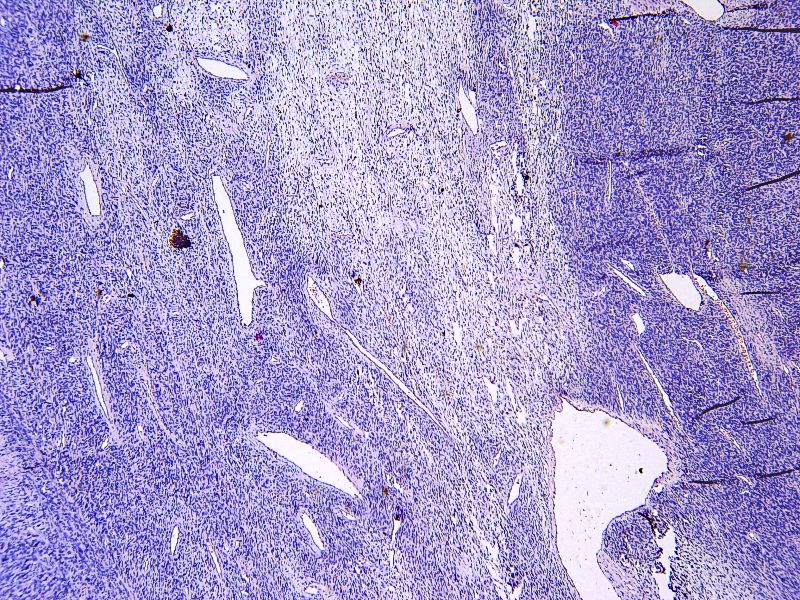
En cuanto a la histología tumoral, se trata de una proliferación de células de hábito fibroblástico, fusiformes u ovales, que se encuentran dispuestas desordenadamente en un tejido conectivo. Se alternan áreas ricas en colágeno con otras hipercelulares.
A mayor aumento presenta un patrón sin patrón en el que se alternan áreas fibrosas de baja celularidad, con otras en las que hay mayor número de células fusiformes dispuestas al azar en un estroma colágeno.
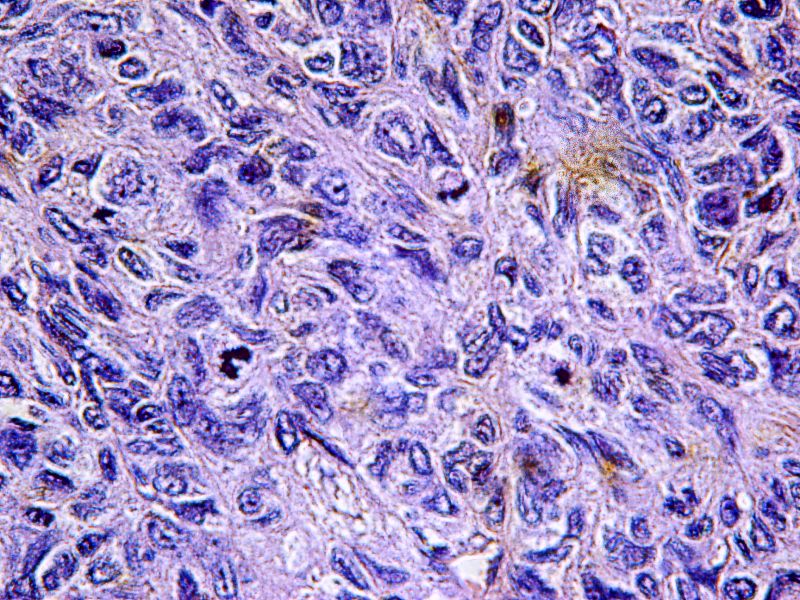
En los cortes seriados del tumor, encontramos zonas donde el tamaño de las células era más heterogéneo con pleomorfismo, atipias y mitosis, como estas que os señalo.
Se llegaron a contar 28 mitosis por 10 campos de gran aumento.
Existían también focos de necrosis.
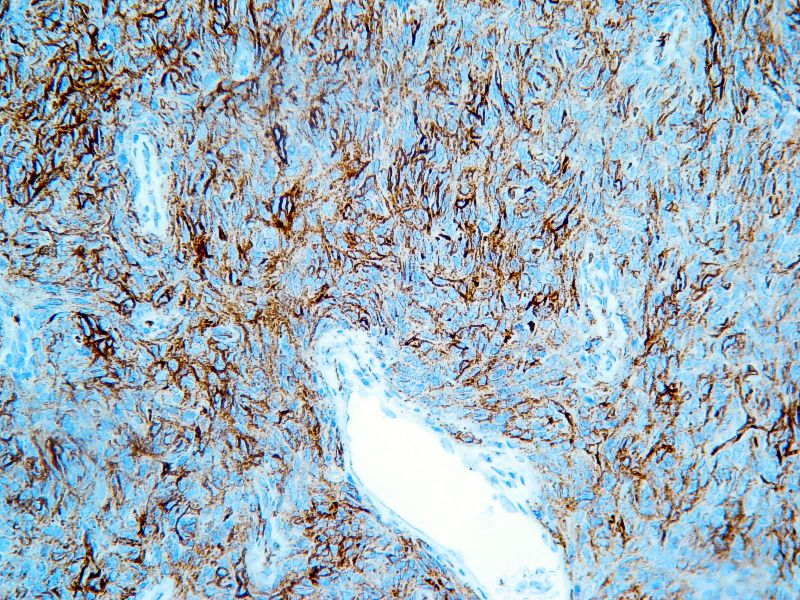
Con la sospecha diagnóstica de Tumor Fibroso Solitario Maligno de la pleura , realizamos técnicas de Inmunohistoquímica La Citoqueratina fue negativa, mientras que el CD34, la Vimentina y el bcl-2 fueron positivos.Este tumor tenía un índice de proliferación celular (Ki-67) del 40%.También se realizó un CD117 (c-Kit) que fue positivo.
El diagnóstico por tanto es de Tumor Fibroso Solitario Maligno de la pleura.
Este es un raro tumor, que supone menos del 5% de los tumores pleurales.
Se da en ambos sexos por igual y a cualquier edad, con más frecuencia entre los 50 y 70 años.
Su localización típica es la pleura viscer al (80% de los casos), pero se han descrito casos localizados en senos paranasales, mediastino, órbita o intrapulmonares.
En general, son tumores que cursan sin síntomas, siendo esto más frecuente en los benignos y son hallazgos casuales en exploraciones por otros motivos. Entre los síntomas que pueden aparecer están la hipoglucemia por secreción de un factor insulina- like y los síntomas locales, tales como el dolor costal, la tos, la disnea o la hemoptisis.
Estas neoplasias suelen presentarse como masas solitarias, bien circunscritas y de tamaño variable. En 2/3 de los casos asientan sobre pleura visceraly más de la mitad son pediculadas.
En cuanto a la histología, como hemos visto en nuestro caso, el patrón sin patrón es la forma más frecuente, con las células fibroblásticas entremezcladas sin orden en un estroma colágeno y con la típicaalternancia de áreas hiper e hipocelulares.
Existen otros patrones como el hemangiopericitoide, el leiomiomatoso, el neurofibromatoso...
La inmunohistoqumica de estos tumores es: la negatividad para la citoqueratina y la positividad para el CD34, fundamentales en el diagnóstico diferencial.
El resto de marcadores de estos tumores que se originan en el mesénquima subpleuralno mesotelial, son la vimentina, el bcl-2 y el CD117 (c-Kit), positivo en el 50% de los casos , y cuyas implicaciones en el tratamiento de este tumor están aún por determinar.
En cuanto a los citerios de malignidad que se describen para este tipo de tumores están: la ausencia de pedículo, la localización atípica (en pleura parietal, mediastino o pulmón), un tamaño superior a 10 cm, que exista necrosis y/o hemorragia y la presencia de células pleomórficas, con atipias y un índice mitótico mayor de 4 mitosis por 10 campos de gran aumento.
El diagnóstico diferencial ha de hacerse con el Mesotelioma sarcomatoide, para lo cual la Inmunohistoquímica es de gran ayuda , y también con tumores malignos de partes blandas tales como el leiomiosarcoma o el sarcoma sinovial monofásico.
En cuanto al tratamiento , es quirúrgico, siendo la extirpación completa la mayor garantía de no recidiva en un futuro.
Tanto la Quimio como la Radioterapia adyuvantes no han demostrado aumentar la supervivencia de estos pacientes.
A pesar de todo esto, existe una tasa de recidiva local del 16%, que se relaciona con la incompleta extirpación del tumor, y hasta un 13% de metástasis a distancia tras la resección.