| . |
Reunión de la Asociación Territorial de
Madrid
14 de Noviembre de 2003
HOSPITAL del NIÑO JESÚS
Departamento de Anatomía
Patológica
Hospital Infantil Universitario del Niño Jesús
Daniel Azorín; Isabel Colmenero; Inmaculada de Prada; Imelda González-Mediero
DERMATOMIOFIBROMA: DESCRIPCIÓN DE DOS CASOS Y
DIAGNÓSTICOS DIFERENCIALES
RESUMEN DEL CASO:
Dos niñas de 10 y 12 años que presentan sendas
tumoraciones subcutáneas de 0.5 y 1 cm respectivamente, no dolorosas
y no adheridas a planos profundos, localizadas en la cara lateral
del cuello y en región cervical posterior respectivamente. Con el
diagnóstico clínico de "dermatofibroma" y "tumor cutáneo"
respectivamente se realiza extirpación de ambas. La histología es
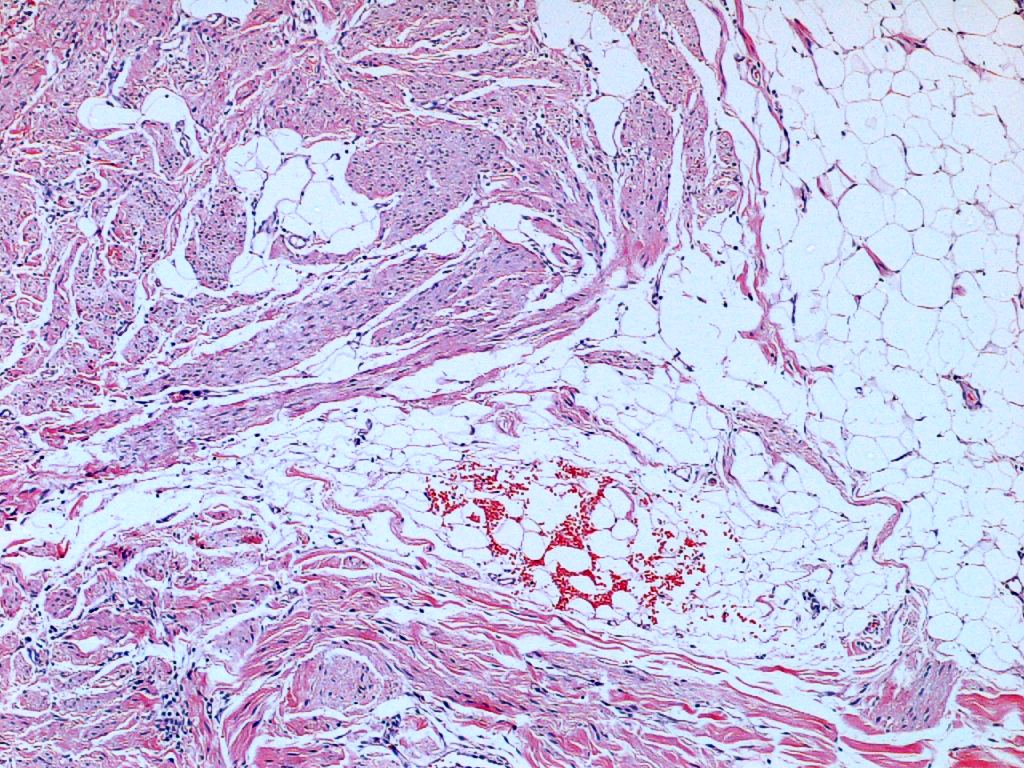
similar en ambos casos. Se trata de una proliferación fusocelular
dispuesta en fascículos paralelos a la epidermis y localizada en
dermis reticular, con afectación superficial del tejido celular
subcutáneo. Dichos fascículos crecen englobando los anejos, sin
destruirlos. No se observan atipias citológicas ni mitosis. Con el
tricrómico de Masson las células muestran características de
fibroblastos/miofibroblastos. La inmunohistoquímica muestra
positividad para vimentina y actina y negatividad para desmina y
S100.

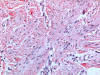
Diagnóstico anatomo-patológico: Dermatomiofibroma
DISCUSIÓN:
El dermatomiofibroma, descrito por primera vez por
Kamino en 1992 (1), constituye una entidad clínico-patológica bien
definida, incluida en el grupo de las lesiones fibroblásticas-miofibroblásticas
de la piel. Existen alrededor de 30 casos descritos en la literatura de
esta entidad. La mayoría de los casos corresponden a mujeres jóvenes,
aunque existen casos descritos en hombres (1, 2,3) y en pacientes en edad
pediátrica (4, 5, 6). En la serie inicial de Kamino (1), que por otro lado
es la más larga, la media de edad estaba en 29.8 años, estando localizados
8 de los 9 casos descritos en la zona del hombro. Otras localizaciones
descritas han sido la región cervical posterior (3, 4, 5, 6, 7), miembro
inferior (1, 2, 3), pared torácica (2, 8, 9) y pared abdominal (6).
Clínicamente la mayoría de los casos son nódulos asintomáticos,
solitarios, de 1-2 cm, aunque existe un caso descrito de 8 cm de diámetro
que se presentó como una lesión lineal (7). En ninguno de los casos
descritos se han observado recidivas locales, ni por supuesto metástasis,
tras un tratamiento quirúrgico conservador en la mayoría de las ocasiones.
Histológicamente (1) se trata de una lesión localizada en la dermis
reticular, adoptando una morfología en placa con extensión superficial al
tejido celular subcutáneo. La epidermis muestra discreta hiperplasia,
observándose un segmento dérmico preservado de forma superior a la lesión.
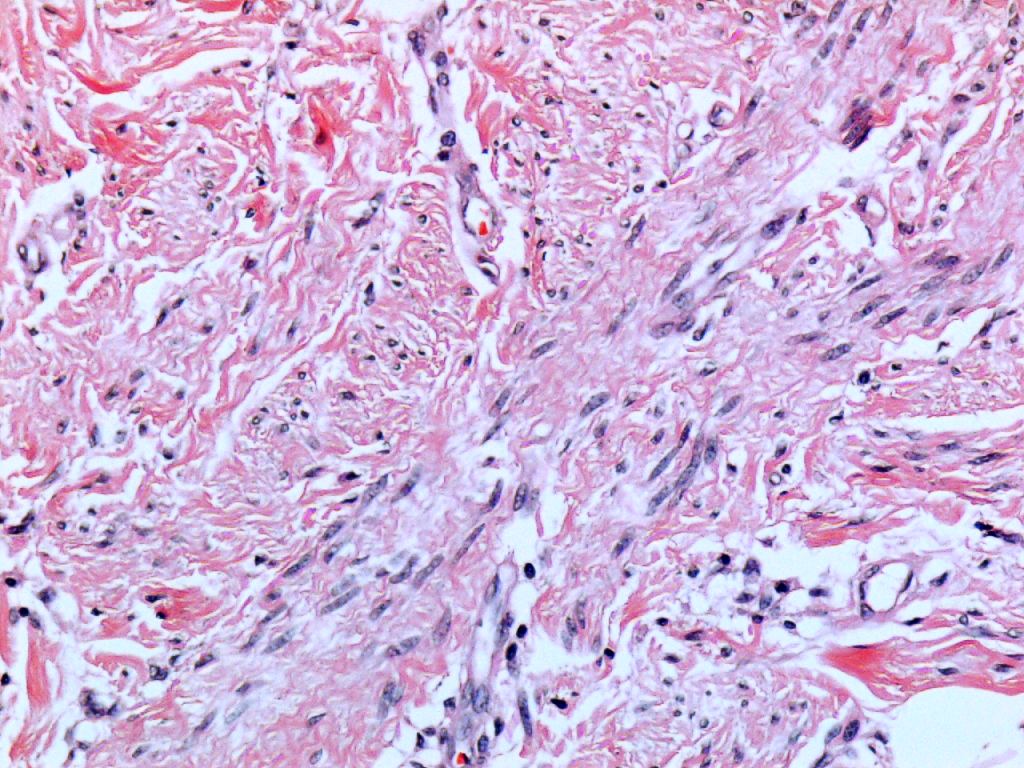
Ésta se encuentra constituida por una proliferación de células fusiformes
monomorfas que constituyen fascículos elongados y entrecruzados, que
adoptan una disposición en paralelo con la superficie epidérmica. Los
fascículos descritos se encuentran rodeando los anejos cutáneos, aunque
sin evidenciarse obliteración de los mismos. Las células fusiformes
presentan núcleos alargados con uno o dos nucleolos, exhibiendo escasa
atipia y escasa o nula actividad mitótica. Con el tricrómico de Masson se
tiñen débilmente de rojo o bien lo hacen de verde, poniendo de manifiesto
su naturaleza fibroblástica-miofibroblástica, en contraste con la tinción
fuscinófila de los músculos erectores del pelo. La tinción para fibras
elásticas de van Gieson revela preservación de éstas entre los fascículos
de la lesión. La inmunohistoquímica confirma la naturaleza de la lesión,
mostrando positividad para vimentina y actina en la mayoría de los casos y
negativas, o débilmente positivas según algunos autores (9), para actina
músculo liso específica. Las tinciones para desmina, S-100, factor XIIIa y
caldesmón (3) son negativas en todos los casos. La microscopía electrónica
colabora en confirmar la naturaleza miofibroblástica de la lesión (8).
En cuanto al origen de esta entidad, la mayoría de los
autores coinciden en su naturaleza neoplásica más que reactiva, basándose
fundamentalmente en la ausencia de historia de traumatismo o cirugía y la
ausencia histológica de sangrado reciente o antiguo, necrosis u otros
cambios degenerativos (1).
El diagnóstico diferencial debe hacerse con otras
lesiones fibroblásticas-miofibroblásticas cutáneas, tales como
dermatofibroma, leiomioma pilar, neurofibroma, miofibromatosis/miofibroma
cutáneo, hamartoma fibroso de la infancia, fibromatosis extra-abdominal y,
por sus especiales implicaciones pronósticas, con el dermatofibrosarcoma
protuberans en placa. El dermatofibroma es una lesión más nodular, con
fascículos dispuestos de forma más desordenada, que destruye los anejos y
que asocia frecuentemente células multinucleadas e hiperpigmentación de la
capa basal de la epidermis. Además la presencia de colágeno queloideo en
la periferia de la lesión y la ausencia de fibras elásticas corroboran el
diagnóstico de dermatofibroma (1). El leiomioma pilar (hamartoma de
músculo liso) presenta una disposición más desordenada y las células
fusiformes poseen núcleos con morfología de cigarro puro. Además es
fácilmente distinguible del dermatomiofibroma con un simple tricrómico de
Masson, ya que las células del primero son fuscinófilas (1). Si fuera
necesario recurrir a la inmunohistoquímica, el leiomioma pilar es positivo
para desmina y caldesmón, ambos negativos en el dermatomiofibroma (3). El
neurofibroma presenta unos núcleos más pequeños y ondulados y es positivo
para S-100, anticuerpo constantemente negativo en el dermatomiofibroma
(6). La miofibromatosis/miofibroma cutáneo presenta un característico
patrón bifásico y una positividad variable para caldesmón, ambos hechos
ausentes en el dermatomiofibroma (3). El hamartoma fibroso de la infancia
es una lesión prácticamente exclusiva de la época prepuberal, siendo muy
característico su patrón organoide constituido por trabéculas de tejido
fibrocolágeno, células primitivas mesenquimales y tejido adiposo maduro,
siendo fácil diferenciarlo del dermatomiofibroma con la simple microscopía
óptica (10). La fibromatosis extra-abdominal es una lesión más profunda,
de mayor tamaño y pobremente circunscrita, mientras que el
dermatomiofibroma es más circunscrito y superficial (1). Aunque es
importante diferenciar el dermatomiofibroma de las entidades anteriormente
mencionadas, no sólo por mero academicismo, sino por la ausente capacidad
de recidivar del primero a diferencia de algunas de estas últimas,
especial hincapié debe hacerse en la diferenciación con el
dermatofibrosarcoma protuberans por sus importantes implicaciones
pronósticas. Este último es más celular, presenta un patrón de crecimiento
estoriforme frente a la disposición en paralelo del dermatomiofibroma,
destruye los anejos cutáneos e infiltra el tejido adiposo de forma
profunda y disecando entre los adipocitos. En casos de biopsias
superficiales, la distinción puede ser muy difícil. En estos supuestos la
positividad para CD34 en el dermatofibrosarcoma protuberans deshace las
dudas (6).
En resumen, el dermatomiofibroma es una entidad clínico
patológica bien definida de origen miofibroblástico que cura totalmente
con extirpación simple, hecho por el que debe ser diferenciado de otros
tumores fibroblásticos/miofibroblásticos, en especial del
dermatofibrosarcoma protuberans.
BIBLIOGRAFÍA:
Kamino H, Reddy VB, Gero M, Greco MA.
Dermatomyofibroma. A benign cutaneous, plaque-like proliferation of
fibroblasts and myofibroblasts in young adults. J Cutan Pathol 1992;
19: 85-93
Mentzel T, Kutzner H. Hemorrhagic dermatomyofibroma
(plaque-like dermal fibromatosis): clinicopathological and
immunohistochemical analysis of three cases resembling plaque-stage
Kaposis sarcoma. Histopathology 2003;42:594-8
DAddario SF, Morgan M, Talley L, Smoller BR. H-Caldesmon
as a specific marker of smooth muscle cell differentiation in some
soft tissue tumors of the skin. J Cutan Pathol 2002; 29: 426-9
Mortimore RJ, Whitehead KJ. Dermatomyofibroma: a
report of two cases, one occurring in a child. Australas J Dermatol
2001; 42: 22-5
Rose C, Broker EB. Dermatomyofibroma: case report
and review. Pediatr Dermatol 1999; 16: 456-9
Mentzel T, Calonje E, Fletcher CD.
Dermatomyofibroma: additional observations on a distinctive cutaneous
myofibroblastic tumour with emphasis on differential diagnosis. Br J
dermatol 1993; 129: 69-73
Trotter MJ, McGregor GI, OConnel JX. Linear
dermatomyofibroma. Clin Exp dermatol 1996; 21: 307-9
NG WK, Cheung MF, MA L. Dermatomyofibroma: further
support of its myofibroblastic nature by electronmicroscopy.
Histopathology 1996; 29: 181-3
Colome MI, Sanchez RL. Dermatomyofibroma: report of
two cases. J Cutan Pathol 1994; 21: 371-6
Hashimoto H. Fibrous hamartoma of infancy. In:
World Health Organization Classifications of Tumours. Pathology and
Genetics of Tumours of Soft Tissue and Bone. Fletcher CDM, Unni KK,
Mertens F, eds. IARC Press: Lyon 2002
|
. |