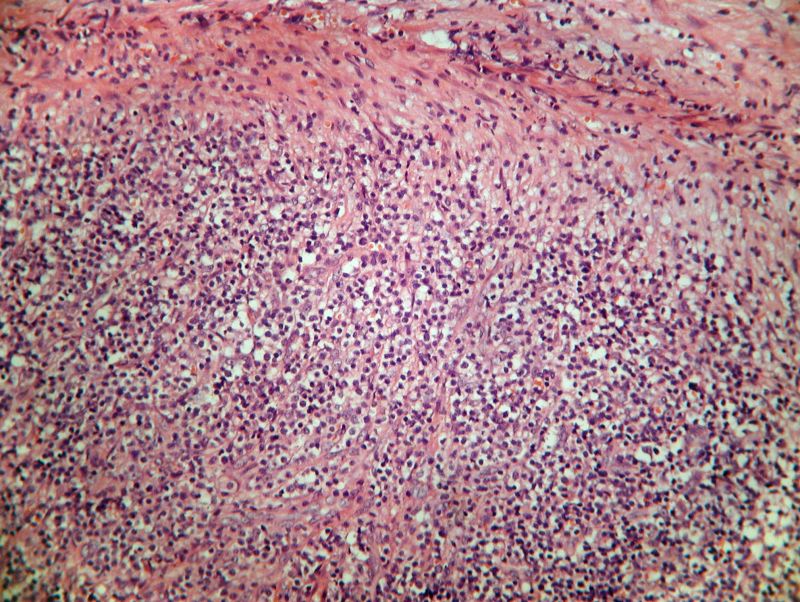
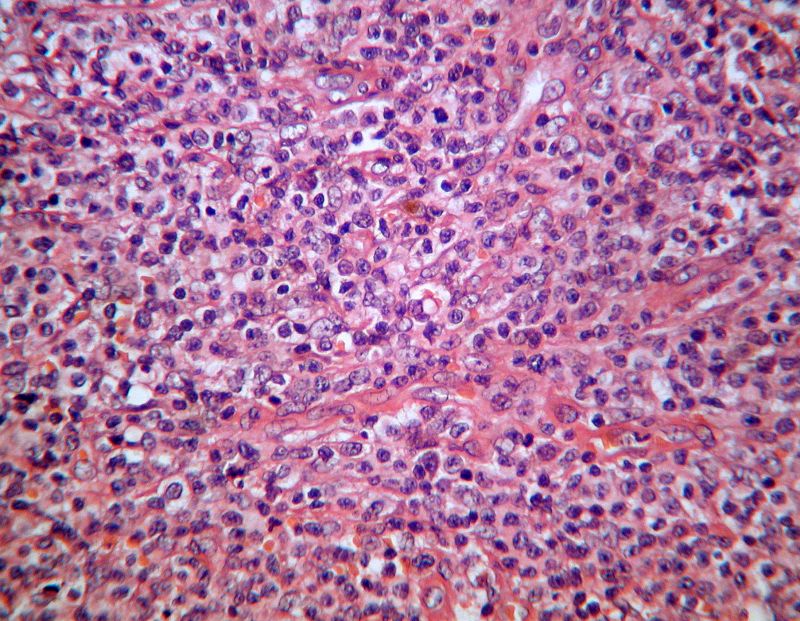
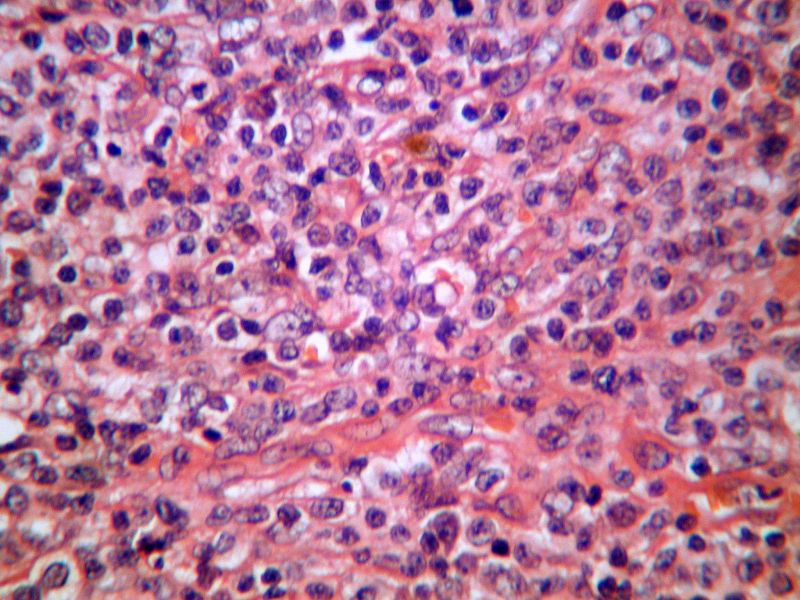
Estudio histopatológico
En el examen microscópico observamos destrucción del
ganglio por una proliferación neoplásica linfoide de patrón difuso
compuesta por elementos intermedios, monomorfos, moderadamente atípicos y
en ocasiones de pequeño tamaño, de núcleo vesiculosos y citoplasma amplio,
dispuestos formando nidos con algunos elementos de núcleo en herradura,
que están entremezclados con una celularidad abundante linfoide de tipo
reactivo. El estroma es ricamente vascularizado. Las mitosis se observan
con facilidad y no se observaron focos de necrosis.
Realizamos estudio inmunohistoquímico sobre material
fijado en formol e incluido en parafina utilizando anticuerpos
monoclonales, con los siguientes resultados:
CD20: negativo. Positividad en menos del 25% de la
celularidad acompañante.
CD79ª: negativo. Positividad en menos del 25% de la
celularidad acompañante.
CD23: negativo.
CD45RO: positividad de membrana fuerte en la
celularidad tumoral y en más del 50% de la celularidad reactiva
acompañante.
CD43: positividad de membrana débil en la celularidad
tumoral y en más del 50% de la celularidad reactiva acompañante.
CD3: negativo en la celularidad tumoral y positivo en
más del 50% de la celularidad reactiva acompañante.
CD30: positividad fuerte de membrana en la celularidad
tumoral con refuerzo paranuclear (Golgi), especialmente en elementos con
disposición perivascular.
P80(ALK): positividad nuclear, nucleolar y
citoplasmática en la celularidad tumoral.
Ciclina D1: negativo
Bcl2: negativo
Ki67 (MIB 1, fracción de proliferación): positividad
nuclear fuerte en el 25-50% de las células tumorales.
P53: negativo
Con estos resultados concluimos que se trataba de un
linfoma anaplásico ALK positivo, variedad histológica de células
pequeñas. El patrón de positividad para p80 era sugestivo de la
presencia de t(2;5) con expresión de NPM-ALK (p80).
Discusión
El linfoma anaplásico de células grandes (ALCL) es un
linfoma de células T constituido generalmente por células grandes con
abundante citoplasma y núcleos pleomorfos (núcleos en herradura). Estas
células son CD30+ y la mayoría expresan proteinas asociadas-gránulo
citotóxicas. La mayoría son también positivas para la proteína quinasa del
linfoma anaplásico (ALK).
Representa aproximadamente el 3% de los linfomas
no-Hodgkin del adulto y el 10-30% de los linfomas en la infancia.
Se reconocen distintas variantes citomorfológicas:
ALCL, variante común (70%). Compuesta
predominantemente por células grandes, pleomórficas, con núcleos
arriñonados o en herradura y una región eosinófila en la región cercana al
núcleo (célula hallmark). También presenta células tumorales más
monomórficas, con núcleos redondos.
ALCL, variante linfohistiocítica (10%). Las células
tumorales se entremezclan con un gran número de histiocitos.
Ocasionalmente estos histiocitos muestran signos de eritrofagocitosis.
ALCL, variante célula pequeña (5-10%). Población
predominante de células neoplásicas pequeñas-intermedias con núcleos
irregulares. Las células hallmark están presentes y se concentran
alrededor de los vasos. Esta variante se confunde con el linfoma T
periférico inespecífico con H&E.
Inmunofenotipo. Las células tumorales son positivas
para CD30 con tinción de membrana y de la región del Golgi (paranuclear).
La inmunotinción es más fuerte en las células de mayor tamaño. En la
variante de célula pequeña también la expresión más fuerte de CD30 es en
las células de mayor tamaño, que suelen presentar agrupamiento vascular.
La expresión de ALK/p80 tiene lugar en el 60-85% de los
casos. La tinción puede ser citoplásmica y nuclear o bien sólo
citoplásmica o sólo nuclear. En la mayoría de los casos, en los que se
observa la traslocación t(2;5)/NPM-ALK, el patrón de tinción es nuclear y
citoplásmica. Este patrón es el resultado de la fusión de una porción de
la proteína ALK (que es una proteína transmembrana) con una proteína
nuclear de transporte (NPM).
La expresión ALK/p80 es específica para ALCL ya que
está ausente en tejidos humanos sanos postnatales excepto aisladas
células cerebrales-. Está también ausente en el resto de neoplasias
linfoides con excepción de ocasionales linfomas B difusos de células
grandes con morfología plasmocitoide. En este pequeño grupo la expresión
de ALK es citoplásmica granular y paranuclear y no se demuestra alteración
genética (además, son CD30- y CD45 débilmente positivo)
Los ALCL negativos para ALK muestran morfología similar
a la variante común pero con mayor pleomorfismo y no suelen mostrar
afectación extranodal. La edad de presentación se eleva y el curso clínico
es más agresivo. No hay estudios genéticos que demuestren si son la misma
entidad.
La gran mayoría de ALCL expresan uno o más antígenos T
(CD3 25%; CD43 65%, CD45 variable%). En los casos con pérdida de expresión
pan T se demuestra expresión T a nivel genético.
También se observa positividad para proteínas asociadas
a gránulos citotóxicos, granzima B y/o perforina.
ALCL no muestran positividad para el virus Epstein-Barr.
Genética. Aproximadamente el 90% de los ALCL
muestran reordenamiento clonal para receptores de células T, expresen o no
inmunfenotipo T.
La expresión de ALK en el ALCL es debida a una
alteración genética del locus ALK en el cromosoma 2. La más frecuente es
la traslocación (2;5) entre el gen ALK del cromosoma 2 y el gen NPM del
cromosoma 5, aunque se han detectado otras (ver tabla).
Pronóstico y factores predictivos. El indicador
pronóstico más importante es la expresión de ALK, que se asocia con
favorable pronóstico. No se han observado diferencias entre tumores con
expresión t(2;5)ALK/NPM y tumores con otro tipo de traslocación. El índice
de supervivencia a 5 años es del 80% en ALCL ALK+ y de 40% en ALK-.
Bibliografía
Pittaluga S et al. Am J Surg Pathol 1997; 1151(2):
343-351
Pulford K et al. Blood 1997; 89(4): 1394-1404
Downing JR et al. Blood 1995; 85(12): 3416-3422
Kinney MC et al. AM J Surg Pathol 1993; 17: 859-868
Benharroch D et al. Blood 1998; 91: 2076-2084
Jaffe ES and Harris NL. WHO Classification of tumours:
Pathology and genetics tumours of haematopoietic and lymphoid tissues.
IARC Press:lyon 2001