| . |
Reunión de la Asociación Territorial de
Madrid
15 de DICIEMBRE de 2001
HOSPITAL DE MÓSTOLES
Hospital Virgen de la
Salud. Complejo Hospitalario de Toledo
Gerardo Pérez Bautista, Manuela
Mollejo Villanueva, Juan Luis Orradre Romeo, Esperanza Carabias López,
Rufo Rodríguez Merlo
HISTORIA CLÍNICA
Niña de 10 años que ingresa en el Servicio de Pediatría
por un cuadro de 15 días de evolución de cefalea frontal progresiva con
características de organicidad acompañada de vómitos aunque sin
objetivarse fiebre. En TAC craneal se apreció masa en sustancia blanca
profunda frontal derecha de 5X 5x4,5cm.
Se procedió a intervención quirúrgica apreciándose una
tumoración rojo-grisácea que se diferenciaba perfectamente de la sustancia
blanca adyacente, realizándose extirpación completa.
ESTUDIO ANATOMO-PATOLÓGICO
Macroscopicamente, se recibieron varios fragmentos de
tejido de coloración grisácea y consistencia blanda que agrupados medían 4
x 3 x 1 cm.
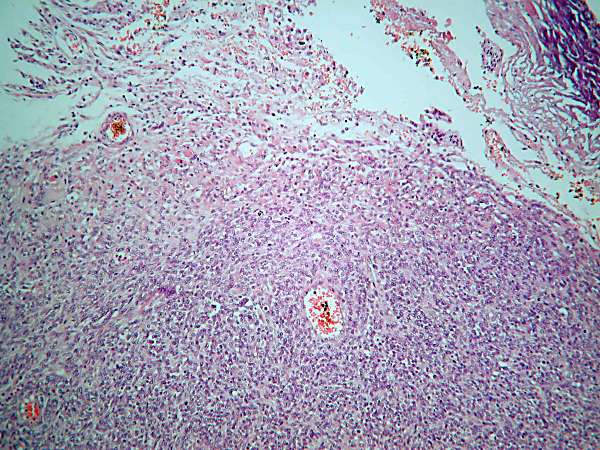
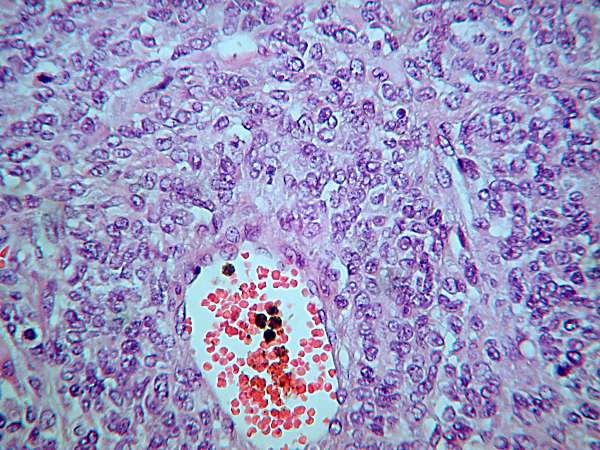
En los cortes histológicos se observa una proliferación
neoplásica de células de pequeño tamaño, muchas de ellas fusiformes, con
abundantes mitosis y focos de necrosis. Ocasionalmente se observa la
presencia de células de núcleo excéntrico y amplio citoplasma eosinófilo
sugestivas de rabdomioblastos, así como presencia de células gigantes
multinucleadas.
En el estudio inmunohistoquímico, la neoplasia muestra
positividad para vimentina, desmina (generalizada e intensa en
rabdomioblastos y células indiferenciadas), y actina focalmente. Presenta
negatividad para queratinas, antígeno común leucocitario, proteína glial
fibrilar, proteína S-100, sinaptofisina, cromogranina y CD99. El índice
proliferativo es intermedio-alto.
Se realizó estudio de la neoplasia mediante FISH para
descartar traslocación t(11;12), no objetivándose dicha alteración.
Diagnóstico: Rabdomiosarcoma
embrionario cerebral
DISCUSIÓN
El rabdomiosarcoma cerebral, tanto primario como
metastásico, es un tumor muy poco frecuente, descrito tanto en
leptomeninges como en parénquima cerebral, de curso clínico agresivo y mal
pronóstico.
Se trata de tumores malignos de células pequeñas,
caracterizados por mostrar diferenciación hacia músculo esquelético. La
citología tumoral está compuesta por un número variable de células
redondeadas, ovales, poligonales o fusiformes. El grado de diferenciación
rabdomioblástica es variable.
En el estudio inmunohistoquímico, muestra positividad
con desmina, actina músculo-específico y MyoD1. Solo ocasionalmente se
puede observar positividad focal o débil con proteína S-100, queratinas,
CD57, y CD99.
La alteración molecular descrita en los
rabdomiosarcomas embrionarios es deleción del brazo corto del cromosoma
11.
El diagnóstico diferencial más importante que se
plantea en el caso que presentamos es el tumor neuroectodérmico primitivo
(PNET), puesto que no es raro encontrar en estos tumores diferenciación
rabdomioblástica, y son neoplasias cerebrales más frecuentes en niños que
el rabdomiosarcoma. Aunque en los PNET se pueden encontrar células
positivas para desmina, la intensa y generalizada positividad de este
grupo de tumores con CD99 ayuda al diagnóstico diferencial. Además, la
detección de la translocación t(11;22) propia de los PNET establece el
diagnóstico. En nuestro caso, la negatividad con CD45, queratina,
cromogranina, sinaptofisina, CD99, S-100, HMB45 y EMA descartaron otros
tumores (linfoma/leucemia, tumor rabdoide, PNET, meningioma, neuroblastoma,
metástasis de melanoma o de nefroblastoma). Por otro lado, la ausencia
tanto de positividad con CD99, como de la traslocación t(11;22) (realizada
en el Centro Nacional de Investigaciones Oncológicas), descartan un PNET.
Clínica y radiológicamente no se ha encontrado un
posible tumor primario extracraneal, por lo que asumimos el posible origen
primitivo en cerebro.
|
. |