Mª Jesús Fernández-Aceñero,
Luis Cortés Lambea
HOSPITAL DE MÓSTOLES
Caso clínico:
varón de raza negra de 25 años natural de Guinea, que
consulta por presentar desde noviembre de 2000 una masa en el hueco
poplíteo derecho. Sólo tenía antecedentes de paludismo y refería una
ligera pérdida de peso en relación con el episodio actual.
Imagen radiológica:
masa de gran tamaño lítica en el tercio distal del
fémur, que respeta el espacio articular de la rodilla y se acompaña de una
gran masa de partes blandas. Los diagnósticos radiológicos más probables
fueron quiste hidatídico óseo o quiste óseo aneurismático.
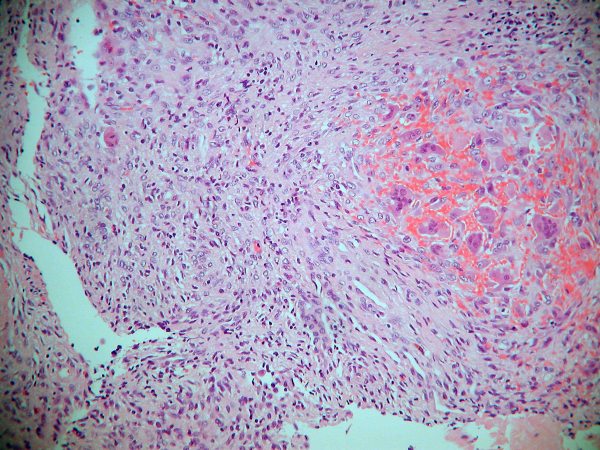
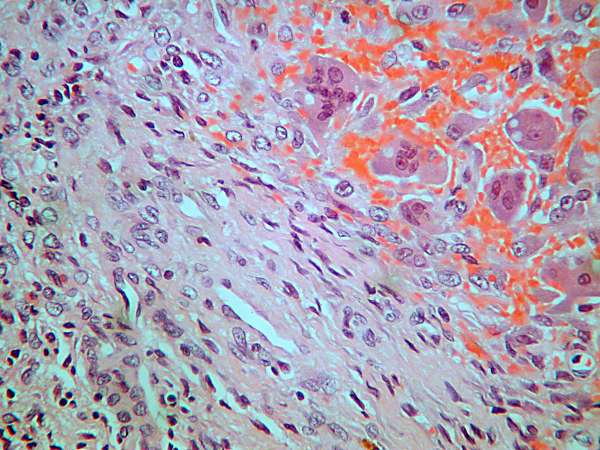
Hallazgos morfológicos:
se resecó una masa de 14 cm de diámetro, que al
corte correspondía a un quiste trilocular con áreas carnosas blanquecinas
en la pared. El estudio de las mismas muestra una proliferación de células
fusiformes de límites mal definidos y citoplasmas eosinófilos, que
presentan leve pleomorfismo y muy escasas mitosis (2/10 CGA). No se
reconoce patrón en espina de pescado ni tampoco estoriforme. No se
observan áreas de necrosis. Hay ocasionales áreas de tipo quiste óseo
aneurismático y acúmulos de polinucleares. Con inmunohistoquímica sólo se
objetiva positividad para vimentina, siendo desmina, HHF35, S100, CD99,
CD34 y los marcadores epiteliales negativos.
Consultado el
caso con el Dr. Fletcher en Boston, hizo el diagnóstico de sarcoma
miofibroblástico de bajo grado al haber obtenido positividad de las
células para a-actina y vimentina.
Posteriormente se procedió a la
amputación del miembro inferior derecho (a los 3 meses del diagnóstico
inicial), observando recrecimiento del tumor en el lugar de la cirugía
previa. El paciente sigue asintomático y libre de enfermedad.
Discusión:
El miofibroblasto es una célula que comparte
características ultraestructurales con el fibroblasto y las células
musculares lisas (1). Aunque se conoce desde hace tiempo su importancia en
algunas funciones fisiológicas (cicatrización de las heridas) (2) y su
presencia en algunos tumores mesenquimales, tanto benignos como malignos (fibromatosis),
sólo recientemente se han empezado a describir tumores constituidos de
forma predominante por este tipo celular (miofibroma cutáneo,
miofibroblastoma de mama) (3,4). El tumor maligno de bajo grado originado
en los miofibroblastos (sarcoma miofibroblástico) fue descrito como
entidad en 1998 por Mentzel y Fletcher, en una serie de 18 casos de
distintas localizaciones (5). Este tumor está constituido por una
proliferación de células mesenquimales, que crecen adoptando un patrón
fascicular y que muestran características inmunohistoquímicas de
miofibroblastos (positividad con a-actina, desmina y/o actina HHF35).
Suelen ser lesiones con baja actividad mitótica, sin necrosis y que pueden
mostrar agregados inflamatorios (polinucleares). El diagnóstico
diferencial se plantea con las fibromatosis, con los fibrosarcomas
clásicos e inflamatorios y, en nuestro caso, también con tumores primarios
óseos como el osteosarcoma de tipo fibroma desmoplásico. El comportamiento
de la mayoría de estas lesiones ha sido bueno, con escasa tendencia a la
recidiva. Recientemente se ha publicado una serie de 4 casos de
miofibrosarcomas óseos, 1 de los cuales falleció por la enfermedad (6).
Bibliografía:
Schurch W, Seemayer TA, Gabbiani
G. The myofibroblast. A quarter century after its discovery Am J Surg
Pathol 1998; 22: 144-147.
Gabbiani G The cellular
derivation and the life span of the myofibroblast. Path Res Pract
1996; 192: 708-711.
Mentzel T, Fletcher CDM The
emerging role of myofibroblasts in soft tissue neoplasia Am J Clin
Pathol 1997; 107: 2-5.
Lee AH, Sworn MJ, Theaker JM,
Fletcher CDM Myofibroblastoma of the breast: a immunohistochemical study
Histopathology 1993; 22: 75-78.
Mentzel T. Dry S, Katenkamp D,
Fletcher CDM Low grade myofibroblastic sarcoma: analysis of 18 cases in
the spectrum of myofibroblastic tumours. Am J Surg Pathol 1998;
22: 1228-1238.
Watanabe K, Ogura G, Tajino J,
Hoshi N, Suzuki T. Myofibrosarcoma of the bone: a clinicopathological
study Am J Surg Pathol 2001; 25:1501-1507.