|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
|
Información || Congresos || Cursos || Territoriales || Noticias || Patología || Telepatología |
| . |
[XXII Congreso Nacional (Palma de Mallorca)] [XXI Congreso Nacional (Madrid)] [ XX Congreso Nacional (Pamplona) ] [XIX Congreso Nacional (Barcelona) ] [XVIII Congreso Nacional (Málaga) ]
CURSO CORTO DE PATOLOGÍA MOLECULAR II Neoplasias linfoides. Marcadores para el diagnóstico y predicción de respuesta. Juan F. García
El importante avance en los conocimientos sobre de la biología celular y molecular del cáncer que en los últimos años hemos podido adquirir está determinado, principalmente, por el uso cada vez más generalizado de los diferentes métodos de análisis molecular masivo. En especial, la posibilidad real de estudiar la expresión génica (RNA o proteínas) de miles de genes simultáneamente en muestras clínicas tumorales está cambiando la perspectiva de los análisis biológicos y sustituyendo el tradicional método monogénico de estudio de las neoplasias. Las dos tecnologías mas ampliamente utilizadas en la actualidad son los arrays de cDNA y la tecnología de tissue-microarray (matrices tisulares). Ejemplo de tissue-microarray, construido con muestras de Linfoma de Hodgkin. Como puede verse, cilindros de 1 mm de diámetro son sobradamente informativos
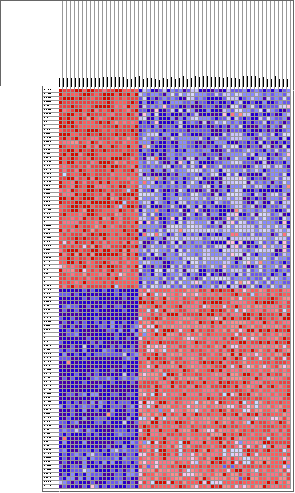
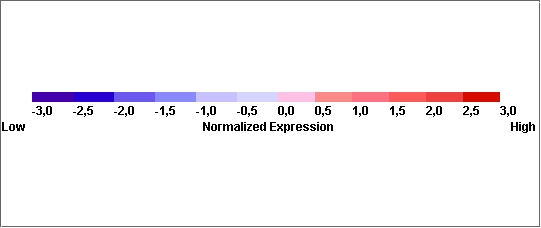
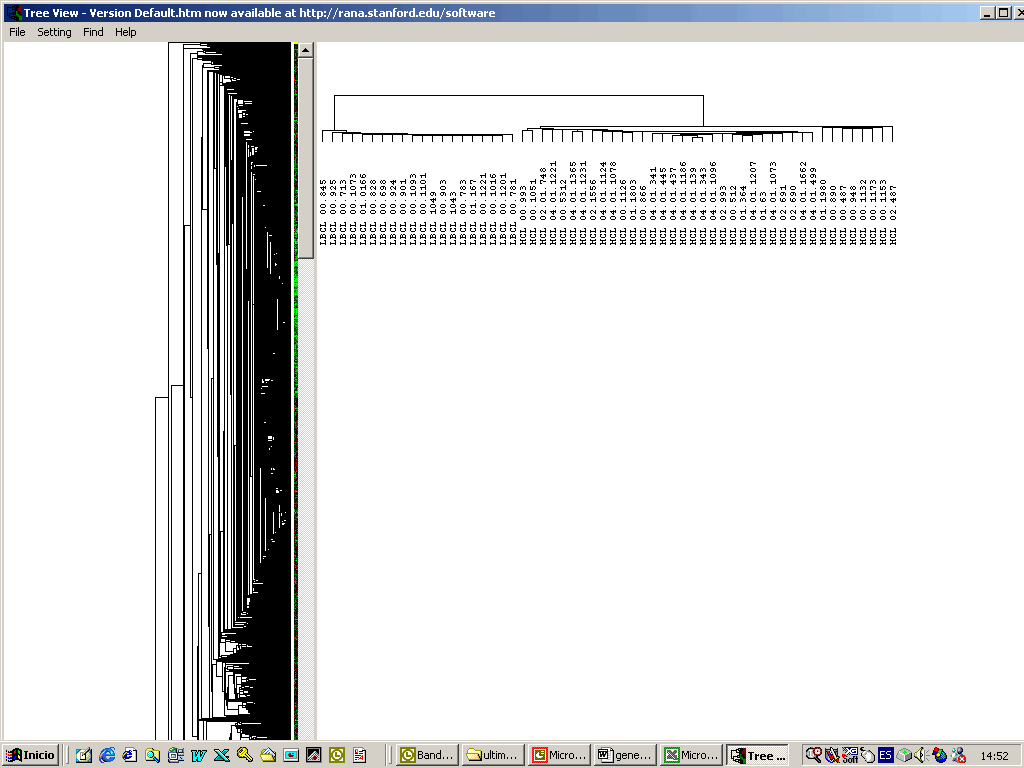
Arrays de cDNA : el mRNA obtenido de la muestra problema, marcado con fluorescencia, se híbrida frente a un mRNA de referencia. El patrón de hibridación es analizado con un escáner basado en microscopía confocal, y la imagen resultante se convierte a valores numéricos. El nivel de expresión de un gen (cuantificación de expresión génica diferencial) se refleja en el número de copias de mRNA y por tanto, es proporcional al nivel de señal detectado. El análisis masivo y simultaneo de la expresión de numerosos genes en chips de cDNA puede aportar la información necesaria para establecer un perfil fenotípico que permita no solo incrementar nuestro conocimiento sobre los mecanismos íntimos de las enfermedades, sino también encontrar una relación entre marcadores biológicos, conducta clínica y respuesta al tratamiento. Los estudios iniciales que se realizaron en linfoma B de células grandes, melanoma y cáncer de mama han mostrado que: La técnica es reproducible y relativamente sencilla. Cada tumor en cada paciente tiene un patrón de expresión propio. Estos patrones se pueden agrupar mediante técnicas bioinformáticas. Los grupos así definidos permiten una correlación con pronóstico, diseminación tumoral y respuesta a fármacos, más allá de la disponible actualmente con técnicas basadas en la exploración clínica o análisis molecular limitado a genes precisos. Distintos Tumores Muestran Diferentes Perfiles de Expresión Génica Se muestra como ejemplo las diferencias que se pueden encontrar al analizar y comparar una serie de 20 linfomas B difusos (LBDCG) y 38 Linfomas de Células del Manto (LCM). Se encuentran más de 2900 genes diferencialmente expresados (p<0.001, test t-Student).
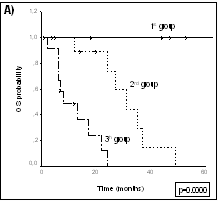
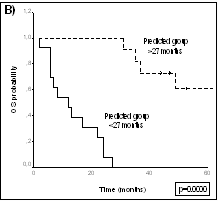
La posibilidad de diferenciar los distintos tumores en base a perfiles de expresión hace de esta tecnología una potencial y prometedora herramienta de diagnóstico. En un estudio independiente realizado sobre linfomas cutáneos de células T, se han encontrado 34 genes diferencialmente expresados (p<0.001) entre los casos de Micosis Fungoides y procesos inflamatorios cutáneos. Este conjunto de genes nos permite la construcción de un modelo predictor que muestra un índice de acierto superior al 90% tras validación cruzada y en el grupo control independiente de pacientes. Modelos Predictores de Supervivencia Una de las principales y más prometedoras aplicaciones de la tecnología de cDNA Arrays es la posibilidad de predecir el curso clínico de los tumores a partir de los datos de expresión génica obtenidos. Teniendo en cuenta la ingente cantidad de información que puede ser generada en este tipo de estudios, el reto actual fundamental estriba en el análisis bioinformático de la información para la construcción de modelos predictivos. Con esta finalidad, se están empleando diferentes aproximaciones matemáticas que emplean modificaciones de procedimientos bioestadísticos tradicionales: análisis discriminante, modelos multivariante de Cox, modelos de regresión logística, etc. La figura representa dos diferentes análisis de supervivencia mediante el método de Kaplan-Meier y log-rank, a partir de un modelo multivariante de Cox (panel A) que separa tres categorías de riesgo, o mediante análisis discriminante (panel B) que predice probabilidad de larga supervivencia versus corta supervivencia, en una misma serie de pacientes con linfomas de células del manto. Ambos modelos matemáticos se basan en la expresión génica del mismo conjunto de genes seleccionados a partir del análisis de expresión.
REFERENCIAS - Alizadeh, A. A., M. B. Eisen, et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403(6769): 503-11. - Davis, R. E. and L. M. Staudt (2002). Molecular diagnosis of lymphoid malignancies by gene expression profiling. Curr Opin Hematol 9(4): 333-8. - Eisen, M. B., P. T. Spellman, et al. (1998). Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95(25): 14863-8. - Garcia, J. F., F. I. Camacho, et al. (2003). Hodgkin and Reed-Sternberg cells harbor alterations in the major tumor suppressor pathways and cell-cycle checkpoints: analyses using tissue microarrays. Blood 101(2): 681-9. - Kallioniemi, O. P., U. Wagner, et al. (2001). Tissue microarray technology for high-throughput molecular profiling of cancer. Hum Mol Genet 10(7): 657-62. - Moch, H., P. Schraml, et al. (1999). High-throughput tissue microarray analysis to evaluate genes uncovered by cDNA microarray screening in renal cell carcinoma. Am J Pathol 154(4): 981-6. - Rosenwald, A., G. Wright, et al. (2002). The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 346(25): 1937-47. - Staudt, L. M. (2002). Gene expression profiling of lymphoid malignancies. Annu Rev Med 53: 303-18. - Tracey, L., R. Villuendas, et al. (2002). Identification of Genes Involved in Resistance to Interferon-alpha in Cutaneous T-Cell Lymphoma. Am J Pathol 161(5): 1825-37. - van de Vijver, M. J., Y. D. He, et al. (2002). A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347(25): 1999-2009. - van 't Veer, L. J., H. Dai, et al. (2002). Gene expression profiling predicts clinical outcome of breast cancer. Nature 415(6871): 530-6. - Wang, E., L. D. Miller, et al. (2002). Prospective
molecular profiling of melanoma metastases suggests classifiers of immune
responsiveness. Cancer Res 62(13): 3581-6. [Programa del Congreso] [Índice del Curso] [Cursos Cortos] [Seminarios]
|
. | |||
| © SEAP. Sociedad Española de Anatomía Patológica | Actualizado: 09/07/2003 |
||||